O coração é formado por duas bombas distintas e, a partir daí, pode-se definir a pequena e a grande circulação.
O lado direito bombeia o sangue para os pulmões, o que caracteriza a pequena circulação e o lado esquerdo bombeia o sangue para os órgãos periféricos, o que caracteriza a grande circulação.
O sangue que vem da cabeça e dos membros superiores (braços) chega ao coração, pela veia cava superior e o sangue que vem do tronco e dos membros inferiores (pernas) chega pela veia cava inferior.

Ilustração por Shepherd Jesse
As duas veias cavas (superior e inferior) desembocam no átrio direito, já no coração.
O sangue sai do átrio direito, passa pela valva tricúspide (que possui três folhetos/como se fossem “portinhas”) e chega a uma cavidade, chamada ventrículo direito.
Do ventrículo direito, o sangue passa pela valva pulmonar e chega à artéria pulmonar.
Essa artéria divide-se em duas (uma que vai levar o sangue para o pulmão direito e outra que vai para o pulmão esquerdo) e chega aos pulmões.
Nos pulmões, o sangue vai receber o oxigênio e eliminar o gás carbônico, liberando-o para o ambiente.
Depois do sangue já ter recebido o oxigênio, ele é coletado pelas veias pulmonares (que são quatro) e ele é transportado até chegar a uma cavidade chamada átrio esquerdo.
Já no átrio esquerdo, o sangue irá passar pela valva mitral (que possui dois folhetos/“portinhas”), cai no ventrículo esquerdo e ele manda o sangue para o arco aórtico, a fim de chegar a todos os tecidos do corpo.

Por Natasha de Souza & Dra. Vanessa Guimarães – CREMESP 118.641
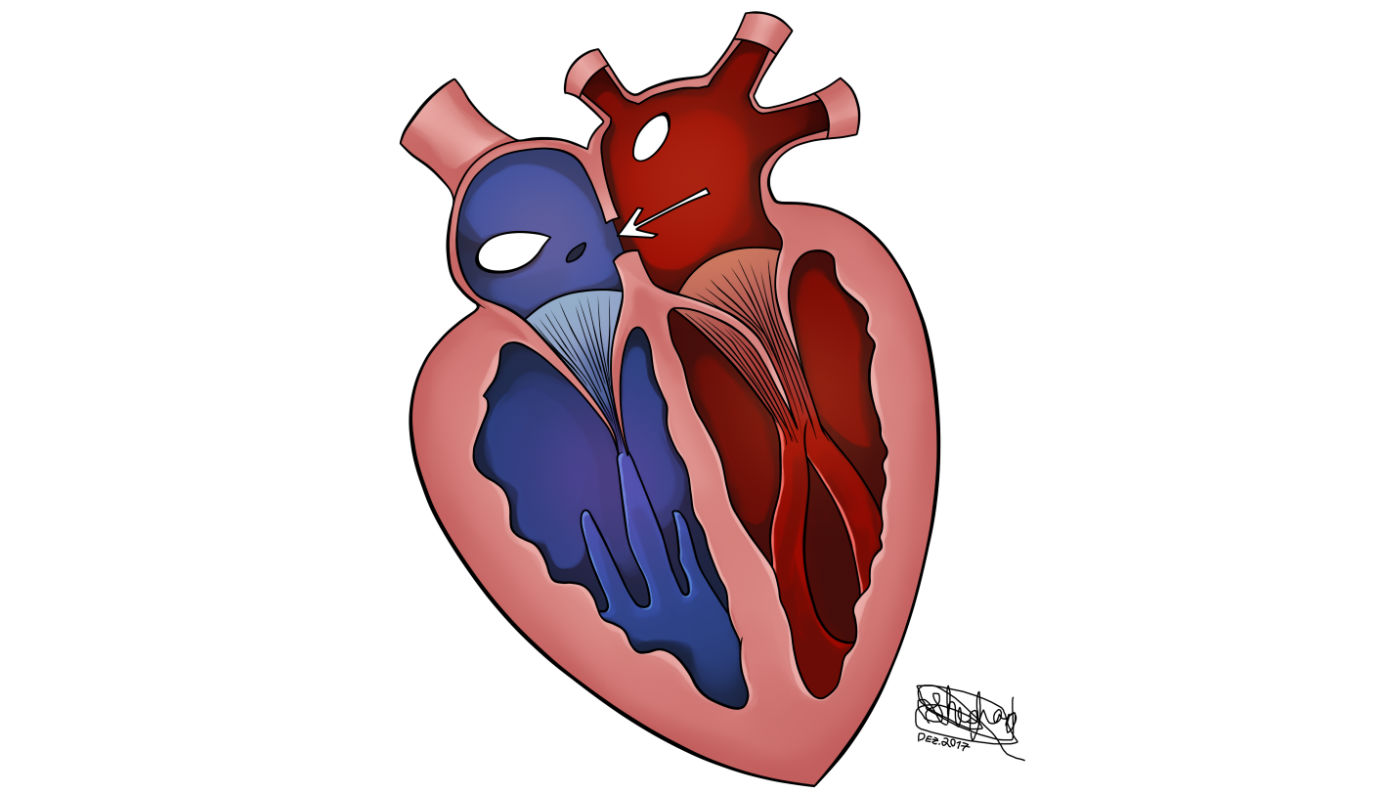
Prevalência na população
Acomete mais de 3 em cada 1.000 nascidos vivos. A prevalência no sexo feminino é o dobro da no masculino. Em sua forma isolada, ela representa 10% das cardiopatias congênitas. Em associação com outras alterações, ela pode chegar a 33% dos casos.
Faixa etária
Presente ao nascimento. O diagnóstico, porém, pode acontecer em qualquer fase da vida.
O que é a doença
Defeito no septo que separa as duas câmaras superiores do coração, os átrios, permitindo a ocorrência de um desvio (shunt) da circulação do átrio esquerdo para o direito. A anomalia gera uma sobrecarga de volume de sangue das cavidades direitas, acarretando um hiperfluxo sanguíneo para os pulmões.
Sintomas
Durante a vida fetal: não há alterações.
No nascimento: sem sintomas, porém, se o defeito no septo dos átrios for amplo e/ou estiver em associação com drenagem anômala das veias pulmonares, pode acarretar taquicardia (batimentos acelerados do coração), dispneia (falta de ar), dificuldade para ganhar peso e sinais de ineficiência de o coração bombear sangue para o organismo (insuficiência cardíaca).
No adulto: falta de ar aos esforços (dispneia), palpitações e cansaço.
Como é diagnosticada
Fetal: A presença do forame oval pérvio (comunicação entre os átrios que é normal no feto) pode dificultar o diagnóstico.
Após o nascimento: sintomas de insuficiência cardíaca (batimentos acelerados e/ou descompassados, falta de ar, dificuldade para ganhar peso) e presença de sopro (som característico dessa malformação) na ausculta com estetoscópio.
No adulto: ausculta cardíaca com sopro típico. Além das alterações percebidas no exame físico, são utilizados exames como radiografia de tórax, que mostra aumento do coração e da trama vascular pulmonar. O eletrocardiograma também mostra-se alterado, nessas circunstâncias (ritmo sinusal e/ou fibrilação e/ou flutter atrial, além de sobrecarga das câmeras direitas e alargamento do intervalo PR).
O ecocardiograma transesofágico é considerado o exame mais indicado para o diagnóstico preciso da comunicação interatrial. Ele permite definir a localização, o tamanho e o número de orifícios, a direção do fluxo, o grau de dilatação das cavidades direitas e a repercussão hemodinâmica.
O que causa a malformação
Nem sempre é possível definir a causa da comunicação interatrial. Alguns casos são genéticos e associados a síndromes: síndrome de Down (10%), de Holt-Oram (autossômica dominante), de Ellis-van-Creveld (autossômica recessiva). Alguns casos são familiares (mutação – genes NKX 2,5 e GATA 4).
Classificação
Cerca de 70% dos casos são o tipo ostium secundum e 20%, ostium primum. Além dessas, existem os tipos menos comuns: tipo seio venoso superior ou seio venoso inferior e tipo seio coronário.
Cuidados
Quando a comunicação interatrial provoca alterações clínicas, gerando dilatação do coração, por exemplo, pode haver a necessidade de restrição de atividades físicas e uso de medicações anticongestionavas até a correção da CIA.
Prevenção
Não há como prevenir a CIA, entretanto, o aconselhamento genético pode identificar o risco de uma cardiopatia congênita vir a ocorrer nas futuras gerações de um indivíduo.
Tratamento
Pode ocorrer fechamento espontâneo da comunicação entre os átrios até 1 ano de idade. Em crianças maiores ou menores muito sintomáticas, deve ser feito tratamento cirúrgico ou percutâneo, para fechamento da comunicação.
Evolução
Sem correção: pode evoluir para insuficiência cardíaca, arritmias atriais, eventos tromboembólicos (formação de coágulos no sangue) e hipertensão pulmonar.
Evolução pós-operatória: excelente.
Com a correção cirúrgica: melhora da performance cardíaca (classe funcional) e redução do tamanho do coração, quando esse sofreu aumento. A correção cirúrgica, no entanto, não diminui o risco de arritmias atriais futuras.
Cura
A correção cirúrgica ou por cateterismo fecha a comunicação interatrial, sem evitar, contudo, o surgimento de arritmias atriais, com o passar do tempo.
Convivência com o problema
É possível conviver com a CIA se o seu diâmetro for pequeno e o aumento do fluxo pulmonar, menor do que uma vez e meia o fluxo sistêmico. Caso a repercussão circulatória seja importante, acarretando sintomas clínicos relevantes, o recomendável é a correção cirúrgica ou por cateterismo.
Prevalência na população
A CIV é uma das mais frequentes entre as malformações cardíacas. Em 20% dos casos em que é detectada, ela aparece de forma isolada, ou seja, não relacionada a outras malformações do coração. Sua incidência chega a alcançar 3,5em cada 1000 nascidos vivos a termo e até 7 em 1000 prematuros. Predomina no sexo feminino(56%).
Faixa etária
Presente ao nascimento, porém o diagnóstico pode ser em qualquer fase da vida.
O que é a doença?
É uma descontinuidade do septo que divide os ventrículos direito e esquerdo.
Sintomas
Vida fetal: nessa fase, as alterações não são detectadas.
Ao nascer: sem sintomas, se ela for moderada, ou, se ampla, aparecem, nos primeiros meses de vida, sinais de insuficiência cardíaca congestiva: taquicardia (ritmo do coração descompassado), taquipneia/dispneia (respiração acelerada/falta de ar), cardiomegalia (coração aumentado), hepatomegalia (aumento anormal do fígado), estertores pulmonares (sons anormais na ausculta do pulmão), palidez da pele, recusa alimentar, baixo ganho de peso, suor excessivo, oligúria (baixa produção de urina) e inchaço nas pernas. Costuma ocorrer também deformidade no tórax da criança.
Como é diagnósticada
Além dos sintomas descritos, observa-se, na ausculta do coração, som bastante característico da anomalia (sopro holossistólicode alta frequência e com intensidade máxima no terceiro e quarto espaços intercostais). Quanto aos exames complementares, utilizam-se o raio X, o eletrocardiograma e o ecocardiograma.
O que causa a malformação
A causa é normalmente desconhecida, entretanto existem estudos que mostram haver correlação entre fatores genéticos e ambientais e também relação entre idade gestacional e peso dos recém-nascidos.
Cuidados
Quando a comunicação interventricular apresenta sintomas, o cardiopediatra orienta a restrição de atividades físicas e o uso de medicações anticongestivas, até que o problema seja corrigido.
Prevenção
Não há como prevenir a CIV. Entretanto, o aconselhamento genético pode identificar o risco de uma cardiopatia congênita vir a ocorrer nas futuras gerações de um indivíduo.
Tratamento
Normalmente são indicadas medicações que ajudam a reduzir a congestão pulmonar, antes da operação. A correção cirúrgica é indicada para a grande maioria dos casos, mas, para casos específicos, o cateterismo também pode ser empregado com bons resultados.
Evolução
A correção do problema pode acontecer de forma espontânea em 5% das CIVs grandes e moderadas e em até em 50% das menores. Nesses casos, no entanto, podem ocorrer aumento da musculatura abaixo da valva pulmonar (hipertrofia infundibular). Devido ao aumento de fluxo sanguíneo para os pulmões, se não corrigida, pode evoluir para hipertensão arterial pulmonar.
Cura
Embora a correção cirúrgica ou por cateterismo possam resolver a comunicação interventricular, pode surgir arritmias, ao longo do tempo.
Como conviver com o problema
Se o diâmetro da comunicação interventricular for pequeno, e a relação entre o fluxo pulmonar e o sistêmico for menor que 1,5:1,0, é possível conviver com o problema sem grandes intercorrências. Caso a repercussão circulatória seja importante, o paciente apresentará sintomas mais severos e, dessa forma, precisará de cirurgia ou de cateterismo para corrigir a malformação. Com ou sem correção cirúrgica, é necessário que o paciente faça prevenção contra a endocardite infecciosa, que é uma infecção por bactéria em uma ou mais das estruturas do coração que tem incidência importante na CIV.
O Defeito de Septo Atrioventricular (DSAV) representa 3 a 4% das cardiopatias congênitas e pode ser dividido em total, parcial e intermediário. Embora esteja presente já ao nascimento, o diagnóstico pode ser feito em qualquer idade.
Também pode ser chamado de Canal Atrioventricular, pois durante a formação do coração ocorre falha nos coxins endomiocárdicos e a região mais central do coração fica comprometida e modificada.
Vamos aqui entender sobre o Defeito de Septo Atrioventricular Parcial, o DSAVP. Nesta cardiopatia existe uma abertura na porção inferior do septo interatrial, chamada de Comunicação Interatrial do tipo Ostium Primum, associada a uma fenda na valva que separa o átrio do ventrículo esquerdos, chamada nesse caso de Valva Atrioventricular Esquerda. Essa fenda gera refluxo sanguíneo do ventrículo pro átrio, ou seja, a valva fica insuficiente e não oclui totalmente a passagem de sangue, o que acaba dilatando essas câmaras cardíacas. Essa insuficiência pode ser discreta, moderada ou importante. Pela comunicação interatrial, ocorre também um shunt da esquerda para a direita, que por sua vez gera aumento do fluxo para os pulmões (a que nos referimos como hiperfluxo pulmonar). O retorno de mais volume sanguíneo para o átrio esquerdo promove sobrecarga de volume das câmaras esquerdas, que somada aos efeitos da insuficiência valvar esquerda deixam constantemente os pulmões mais congestos (cheios de sangue), o que a longo prazo geral a hipertensão pulmonar.

Ilustração por Izabella Hanada
Agora que entendemos o que acontece no coração e com o fluxo de sangue, vamos falar sobre sintomas e repercussões para os pacientes.
A maioria dos portadores de DSAVP são assintomáticos e iniciam os sintomas na 3ª e 4ª décadas de vida. Eles aparecem devido à sobrecarga pulmonar e cardíaca e resultam em insuficiência cardíaca congestiva. Os mais comuns são dispnéia (falta de ar) e intolerância para esforços, taquicardia (freqüência cardíaca acelerada), dificuldade de ganho de peso (pois há muito gasto calórico nesta sobrecarga), recusa alimentar, cardiomegalia (coração aumentado), hepatomegalia (aumento anormal do fígado), estertores pulmonares (sons anormais na ausculta do pulmão por estarem cheios de sangue).
O médico durante o exame físico percebe um sopro mesosistólico, ou seja, no meio da sístole, no foco da valva mitral à ausculta cardíaca e nota também o desdobramento amplo da segunda bulha, o “tac” do “tum-tac” passa a ser ouvido como “tarac”.
Normalmente são necessários exames complementares: – A Radiografia de tórax mostra aumento da área cardíaca e da trama vascular dos pulmões. – O Eletrocardiograma pode apresentar distúrbios da condução elétrica como um bloqueio atrioventricular (BAV) de 1º grau (que é a lentificação da passagem do estímulo elétrico dos átrios para os ventrículos). Além disso, há desvio do eixo elétrico para esquerda (-30º a -90º) e atraso da despolarização ventricular direita como reflexo da sobrecarga de volume. – O Ecocardiograma permite a avaliação da anatomia da malformação descrita e suas repercussões hemodinâmicas (quantificar o grau de insuficiência da valva atrioventricular esquerda – VAVE, o grau de dilatação das câmaras esquerdas e a avaliação da pressão pulmonar).

Ilustração por Izabella Hanada
Quando o DSAVP começa a gerar sintomas, o cardiopediatra reorienta e restringe as atividades físicas e inicia medicações anticongestivas (diuréticos e medicamentos vasodilatadores, que reduzem a pressão arterial e ajudam a diminuir o grau de insuficiência valvar e a congestão pulmonar) até a cirurgia.
É recomendado o aconselhamento genético e o eco fetal para diagnóstico pré-natal.
Essa cardiopatia necessita de correção cirúrgica, pois não ocorre o fechamento espontâneo do defeito. Ela é geralmente indicada entre 1 e 3 anos, se o paciente tiver pouco ou nenhum sintoma, ou antes se o paciente for sintomático. Caso os sintomas se manifestem, são indicadas medicações anticongestivas até a cirurgia.

Por Beatriz Criniti & Dra. Vanessa Guimarães – CREMESP 118.641
O Defeito de Septo Atrioventricular (DSAV) representa 3 a 4% das cardiopatias congênitas e pode ser dividido em total, parcial e intermediário. Embora esteja presente já ao nascimento, o diagnóstico pode ser feito em qualquer idade.
Também pode ser chamado de Canal Atrioventricular, pois durante a formação do coração ocorre falha nos coxins endomiocárdicos e a região mais central do coração fica comprometida e modificada.
Temos o Defeito de Septo Atrioventricular Total (DSVAT) quando há uma falha na fusão dos coxins endocárdicos.
Com isso o defeito inclui:
1. Defeito do septo interatrial, classificado como Comunicação Interatrial (CIA) Ostium Primum.
2. Uma única valva atrioventricular, com 5 folhetos ou válvulas (“portinhas”), separando as cavidades superiores (átrios) das inferiores do coração (ventrículos) – lembrando que o comum é termos 2 valvas, uma do lado direito (a valva tricúspide) e outra do lado esquerdo (a valva mitral);
3. Defeito do septo interventricular, que é classificado como Comunicação Interventricular (CIV) Perimembranosa de Via de Entrada (isso porque ela fica próxima à valva atrioventricular, que é a via de entrada de sangue nos ventrículos.

Prevalência na população Os defeitos do septo atrioventricular total e o parcial, representam 5% dos defeitos congênitos do coração. E, de todos o casos de DSAVT, 30% tem Síndrome de Down.
Está presente já ao nascimento, mas pode ser descoberta em diferentes etapas da vida a depender da gravidade dos sintomas e achados no exame físico.
Sintomas O DSAVT é uma cardiopatia classificada como acianótica, com aumento de fluxo ou hiperfluxo pulmonar, que gera sintomas de insuficiência cardíaca congestiva (ICC) e provoca sintomas como respiração acelerada; falta de ar enquanto o bebê mama ou crianças fazem refeições ou esforços físicos; baixo ganho de peso devido ao consumo energético aumentado pela própria falta de ar; e sudorese, que geralmente se apresenta entre 4 e 6 semanas de idade. O bebê ou criança pode também apresentar pneumonias de repetição.
Diagnóstico
O diagnóstico pode ser feito já durante o Ecardiograma Fetal ou no teste do coraçãozinho, ainda na maternidade, após as primeiras 24 horas do nascimento do bebê.
O pediatra e o cardiopediatra reconhecem os sintomas de insuficiência cardíaca da história do paciente e no exame físico auscultam sopro cardíaco, que é o ruído devido à mudança do fluxo de sangue no coração, neste caso principalmente secundária à insuficiência da valva atrioventricular única.
Os exames complementares: ECG, radiografia de tórax e ecocardiograma ajudam tanto a mostrar a repercussão da cardiopatia no coração e pulmões do paciente, como acompanhar sua progressão.
Tratamento
É cirúrgico, sendo preconizada a correção total do defeito, com o fechamento da CIA, a plástica da Valva Atrioventricular separando-a em direita e esquerda e o fechamento da CIV.
Em pacientes com Síndrome de Down e outras síndromes a cirurgia é feita prioritariamente por volta do sexto mês de vida com o objetivo maior de não se permitir o desenvolvimento da doença hipertensiva pulmonar. Diuréticos e vasodilatadores ajudam a aliviar os sintomas de hiperfluxo e congestão pulmonar antes da cirurgia e ajudam na recuperação pós-cirúrgica.
Normalmente quanto mais insuficiente for a valva atrioventricular, mais sintomas tem o paciente e mais trabalhosa é a correção cirúrgica.
Após a cirurgia, se continuar havendo algum grau de insuficiência da valva, agora separada em direita e esquerda, ou até mesmo estenose, o paciente será clinicamente acompanhado e é possível ser necessária reabordagem cirúrgica para nova plástica da valva ou até troca por valva artificial.
Por Milla Sallenave e Dra. Vanessa Guimarães CRM 118.641
DRENAGEM ANÔMALA TOTAL DE VEIAS PULMONARES

Prevalência na população:
A drenagem anômala total de veias pulmonares é uma anomalia congênita rara e corresponde a cerca de 1-2% das cardiopatias congênitas. Sua prevalência na população é cerca de 0,05 para cada 1000 nascidos vivos e ocorre com maior frequência no sexo masculino.
Faixa etária
Como se trata de uma cardiopatia congênita, a drenagem anômala total de veias pulmonares acomete o indivíduo desde a vida intrauterina.
O que é a doença
A drenagem anômala total de veias pulmonares (DATVP) ocorre quando as todas as veias pulmonares drenam para o átrio direito e não para o esquerdo como o habitual. Dessa forma, todo aporte sanguíneo, sistêmico e pulmonar, chega ao lado direito do coração, ocorrendo uma mistura do sangue oxigenado que vem dos pulmões com o sangue pouco oxigenado que vem da circulação sistêmica. Como não há retorno venoso para o átrio esquerdo, essa cardiopatia é dependente de uma comunicação interatrial e o tamanho dessa comunicação irá determinar como será a distribuição do fluxo sanguíneo nos compartimentos cardíacos. Como a quantidade de sangue que chega ao átrio direito é muito significativa, mesmo na presença da CIA, boa parte do sangue passa para o ventrículo direito ocasionando um hiperfluxo pulmonar. A DTVP pode ser classificada em quatro tipos distintos, sendo eles: Tipo supracardíaco, tipo cardíaco, tipo infracardíaco e misto. Além disso, a DATVP pode se apresentar na forma não obstrutiva ou obstrutiva, que é mais comum no tipo infracardíaco, e nesses casos as manifestações clínicas como de IC irão se manifestar de forma mais intensa e precoce.
Sintomas
De forma geral, as manifestações clínicas irão variar se a DATVP se apresenta de forma obstrutiva ou não obstrutiva, isso é, se há ou não estenose pulmonar. Nos pacientes em que não ocorre obstrução e existe um fluxo considerável pela CIA, a doença pode não manifestar sintomas ao nascimento, e somente após um mês de vida começam a aparecer taquipnéia, cansaço na hora das mamadas e baixo ganho ponderal que falam a favor de Insuficiência Cardíaca. Nesses casos, a cianose pode ser imperceptível ou bem discreta. Já quando o paciente apresenta obstrução pulmonar, os sinais de insuficiência cardíaca e a cianose são mais evidentes e a descompensação ocorre logo nos primeiros dias de vida.
Na forma não obstrutiva, ao exame físico percebe-se sinais de edema leve e na ausculta observa-se hiperfonese e desdobramento fixo da 2ª bulha, com possibilidade da 3ª e até a 4ª bulha estarem presentes. O sopro é sistólico do tipo ejetivo e é identificado em foco pulmonar. Em geral, os sintomas de IC começam a se agravar em torno dos 6 meses. Já na forma obstrutiva, é observada cianose central moderada a intensa, o paciente apresenta grande desconforto respiratório e sinais de baixo débito cardíaco, descompensando subitamente. No exame físico, é observado edema periférico e hepatomegalia. Na ausculta cardíaca é possível perceber um ritmo em galope com presença da 3ª bulha, o sopro pode ser discreto ou até mesmo ausente e na ausculta pulmonar são observados crepitações.
Como é diagnosticada
O diagnóstico da drenagem anômala de veias pulmonares é realizado através de exames como eletrocardiograma, que evidencia sinais de sobrecarga ventricular direita, mas especialmente através do ecocardiograma(ECO), que através da tecnologia do fluxo de cores, tem permitido a identificação cada vez mais precoce da doença. Além disso, o ECO possui maior sensibilidade e especificidade para o diagnóstico, pois torna possível a avaliação do local exato de drenagem, se há ou não obstrução e a presença de CIV. Outro exame que ajuda no diagnóstico é a Radiografia de Tórax(RX) que pode mostrar cardiomegalia com hiperfluxo pulmonar. Pode também surgir uma imagem denominada “boneco de neve” que ocorre mais frequentemente em crianças maiores com o tipo supracardíaco.
Além desses exames, pode ser também realizado cateterismo, que é um método mais invasivo e é reservado para casos em que se deseja avaliar a diferença de pressão nas câmaras e também permite identificar a estenose pulmonar em casos de difícil elucidação.
O que causa a má formação
De forma geral, alguns fatores externos podem influenciar no aparecimento de cardiopatias congênitas, como o uso de drogas lícitas e ilícitas, medicamentos, compostos químicos e fatores genéticos. Esses fatores podem predispor a má formação que ocorre ainda na vida intrauterina.
Durante a formação embriológica do coração, a veia pulmonar primitiva se forma como uma evaginação da parede atrial dorsal à esquerda do septo primum. Conforme o átrio vai aumentando de tamanho a veia pulmonar primitiva vai se incorporando à parede atrial esquerda e se divide em seus quatro veias pulmonares. Quando esse processo ocorre de forma diferente e a incorporação não é no átrio esquerdo e sim no direito ou em algum outro vaso sistêmico, ocorre a drenagem anômala total de veias pulmonares.
Classificação
A DATVP é cardiopatia cianogênica com shunt bidirecional, e pode ser classificada em quatro tipos distintos, são eles:
Cuidados
Para crianças portadoras de DATVP quando vão realizar algum procedimento médico ou odontológico é recomendado utilizar antibióticos como profilaxia pelo risco de endocardite.
Prevenção
Diante de um paciente portador dessa cardiopatia é necessário investigar as possíveis causas que podem ter precipitado a má-formação. Quando é de causa genética, o aconselhamento genético é necessário para prevenir a ocorrência de outras cardiopatias congênitas ou até mesmo problemas extra cardíacos. Além disso, é de suma importância alertar sobre o uso de substâncias que podem precipitar esses eventos, como uso de drogas e medicamentos.
Tratamento
O tratamento definitivo consiste no procedimento cirúrgico, mas para que ele seja feito é necessário estabilizar clinicamente o paciente dos sintomas da Insuficiência Cardíaca. Dessa forma, o tratamento clínico consiste em tratar os sintomas de congestão no tipo não obstrutivo e no tipo obstrutivo podemos ofertar oxigênio e diuréticos. Além disso, se o bebê possui edema pulmonar grave é necessário intubação orotraqueal e ventilação com pressão positiva e pode ser necessário realizar uma atriosseptostomia com balão em casos que é preciso aumentar a CIA. O tratamento cirúrgico está indicado para todos os pacientes, naqueles que possuem o grau obstrutivo deve ser realizado o mais precocemente possível, e naqueles com o tipo não obstrutivo pode ser feita de 4 a 12 meses, desde que estabilizados clinicamente. Vale ressaltar, que as técnicas cirúrgicas irão se diferenciar de acordo com os tipos de Drenagem Anômala Total de Veias Pulmonares.
Evolução
O seguimento do paciente é necessário, pois existem casos em que se descobre estenose pulmonar tardiamente. Além disso, o paciente portador de DATVP está mais susceptível à desenvolver quadros de arritmia como as disfunções de nó sinusal.
Cura
O tratamento cirúrgico corrige a DATVP. De forma geral, metade dos pacientes com a forma não obstrutiva e que não foram submetidos à correção cirúrgica conseguem chegar à vida adulta, mas aqueles que possuem a forma obstrutiva tem baixíssima expectativa de vida, em torno de 2 semanas. No entanto, uma complicação ainda comum no pós operatório é a hipertensão pulmonar que aumenta as chances de morbimortalidade.
Convivendo com o problema
É necessária uma rede de apoio multidisciplinar no acompanhamento do paciente e da família, isso inclui não só os profissionais da saúde, bem como a família, escola e sociedade. Normalmente, os pacientes que precisam de algumas restrições são aqueles que tiveram formas mais graves ou que não fizeram a correção precocemente. Já aqueles sem demais complicações conseguem realizar suas atividades sem grandes problemas.
REFERÊNCIAS BIBLIOGRÁFICAS
Por Mabelle Fragoso & Dra. Vanessa Guimarães – CREMESP 118.641
Prevalência na população
A doença afeta 1 em cada 200 a 250 pessoas. Nasce no mundo um bebê com hipercolesterolemia familiar por minuto.
Faixa etária
Por ser uma doença genética, ela está presente desde o nascimento.
O que é a doença
A hipercolesterolemia familiar é uma doença caracterizada por níveis extremamente altos de de LDL colesterol, ou colesterol “ruim”, no sangue. Tal anomalia acelera o processo de envelhecimento do sistema cardiovascular chamado aterosclerose, levando à formação de placas de ateroma (ou gordura) no interior de artérias e veias concomitantemente ao “endurecimento” da parede arterial. Quando uma dessas placas se rompe, o organismo se esforça para reparar o local do rompimento. Esse processo de autorreparação resulta na formação de coágulos sanguíneos ou trombos na região do interior do vaso que já se encontra estreitada pela placa rompida. Nessa condição de estreitamento, o trombo acaba por funcionar como um tampão naquela região do vaso, impedindo que o sangue passe e alimente a região localizada logo depois do “entupimento”. O tecido que não é alimentado de oxigênio e nutrientes, através do sangue, morre paulatinamente. Esse processo se chama infarto. Quando ele acontece no coração, ele é chamado de infarto do miocárdio (músculo cardíaco). No cérebro, ele se chama AVC (acidente vascular cerebral) ou derrame. O surgimento do infarto é bastante precoce em pessoas que têm hipercolesterolemia familiar que não são tratadas.
Sintomas
Xantomas (tumor benigno composto de gordura localizado especialmente nos cotovelos, joelhos, mãos, pés, coxas e glúteos), xantelasmas (pequenos tumores benignos de gordura localizados nas pálpebras), arco corneal (aureola em volta da íris dos olhos), dores abdominais recorrentes e pancreatites.
Como é diagnosticada
Por meio de exame de sangue e exame genético para identificar se a pessoa tem a mutação genética que predispõe à doença. Se um parente têm hipercolesterolemia familiar, há 50% de chance de a criança ter também.
O que causa a doença
A maioria dos casos se deve à mutação no gene do receptor da lipoproteína de baixa densidade (LDL colesterol ou colesterol ruim), resultando em ausência ou disfunção desse receptor nos hepatócitos (células do fígado). Com isso mais LDL colesterol fica livre na circulação, levando ao aceleramento do processo de aterosclerose.
Cuidados
O diagnóstico precoce é essencial para o controle da doença e o retardamento do processo de aterosclerose, por meio do tratamento medicamentoso contínuo e da adoção de hábitos de vida saudáveis: alimentação balanceada, prática de atividade física, controle do peso e dos níveis de pressão arterial e de glicemia, além da privação do cigarro e de bebidas alcoólicas. Trata-se de uma paciente que deve ser acompanhado de perto, e desde cedo, pelo cardiologista pediátrico.
Prevenção
A doença é determinada pela genética, ou seja, se o pai ou a mãe apresentarem a doença, a chance de a criança também tê-la é de 50%. Portanto, o mais indicado nessa situação é o aconselhamento genético. A evolução da doença é prevenida por meio do tratamento medicamentoso contínuo e da adoção de hábitos de vida saudáveis.
Tratamento
O tratamento consiste no uso de medicamentos de controle do colesterol, como as estatinas, ou de impacto no metabolismo de formação do colesterol (PSK9), junto com modificações no estilo de vida.
Evolução
Se não for tratada de modo adequado e continuado, a hipercolesterolemia familiar pode evoluir precocemente para o infarto do miocárdio e para o AVC.
Cura
Embora seja uma doença que, se tratada, tem controle, a hipercolesterolemia familiar não tem cura.
Convivência com o problema
Crianças com hipercolesterolemia familiar devem ter seu peso, crescimento e desenvolvimento monitorados pelo pediatra, cardiologista pediátrico e muitas vezes por especialistas em lipídios.
Prevalência na população
A verdadeira incidência é desconhecida.
Faixa etária
Mulheres gestantes acima de 30 anos.
O que é a doença?
A miocardiopatia periparto é uma doença rara que acomete mulheres, no final da gestação ou até cinco meses após o parto, causando insuficiência cardíaca (incapacidade de o coração bombear o sangue para o organismo com eficiência).
Sintomas
As pacientes relatam principalmente falta de ar (dispneia), que pode se estender ao período noturno, quando a pessoa se deita para dormir (dispnéia paroxística noturna), tosse, com presença de sangue expectorado pelo pulmão (hemoptise).
Como é diagnosticada
Além dos sintomas clínicos característicos, a doença é diagnostica pelo eletrocardiograma, ecocardiograma, radiografia de tórax, cineangiocoronariografia e exame de tecidos internos ao coração (biópsia endomiocárdica).
O que causa a doença
Dentre as possíveis causas estão miocardite (inflamação do coração), predisposição genética, deficiência de selênio e persistência de alterações fisiológicas da gestação, como a hipertrofia ventricular transitória (aumento do ventrículo cardíaco próprio da gestação, com o propósito de levar mais oxigênio e nutriente para o crescimento do bebê).
São fatores de risco idade acima de 30 anos, já ter tido mais de um filho (multiparidade), ter tido filhos gêmeos (gemelaridade), descendência africana, história de pré-eclâmpsia, eclâmpsia e hipertensão pós-parto ou ter sido submetida a terapia de longo prazo (mais de 4 semanas) com agonistas b-adrenérgicos, como terbutalina.
Prevenção
Evitar os possíveis fatores de risco acima descritos.
Tratamento
Para o tratamento são utilizadas medicações anticongestivos, como bloqueadores beta 1 seletivos e digoxina. A terapia imonossupressiva é uma alternativa especificamente na miocardite documentada pela biópsia miocárdica, após duas semanas de terapia padrão mal sucedida. Imunoglobulina intravenosa pode melhorar a FEVE a curto prazo. O balão intraaórtico e o transplante cardíaco são opções para quadros graves que não responderam ao tratamento convencional.
Complicações
As complicações mais frequentes são insuficiência cardíaca, tromboembolismo e arritmias. Os fenômenos tromboembólicos são frequentes pela hipercoagulabilidade da gestação associada à estase sanguínea dentro das câmaras cardíacas, favorecendo o surgimento de trombose intraventricular, sistêmica e pulmonar.
Evolução
O desfecho é variável. Alguns estudos demonstraram que a disfunção ventricular esquerda torna-se persistente em 50% das pacientes, o que pode resultar em uma mortalidade de 85% em 5 anos.
Convivência com o problema
O planejamento familiar é fundamental, porque as pacientes que normalizaram a função ventricular podem apresentar recidiva da doença em gestação subsequente.

Prevalência na população
É a cardiopatia congênita acianogênica mais comum em recém-nascidos prematuros, correspondendo a até 12% dos casos de cardiopatias congênitas, com predomínio no sexo feminino. É mais comum ainda em prematuros com menos de 1 quilo que apresentam malformação do coração. Nesse caso, incidência da PCA chega a 70-80% dos casos.
Em tempo, doenças acioanogênicas são aquelas que, mesmo provocando mistura de sangue arterial (oxigenado) e venoso (não-oxigenado) dentro do coração, não causam diminuição da oxigenação do sangue para o corpo.
Faixa etária
A PCA está presente desde o nascimento, ou seja, não é uma doença adquirida. Seus sintomas, no entanto, variam caso-a-caso, sendo mais evidentes quando o diâmetro do canal arterial é maior. Nesse caso,o diagnóstico acontece mais precocemente.
O que é a doença?
Trata-se de um pequeno canal (pequeno vaso sanguíneo) presente no feto é que é importante para o desenvolvimento e a vida do bebê, nessa fase.
Situada entre a artéria pulmonar e a crossa da aorta, esse canal é responsável por desviar o fluxo do sangue venoso da artéria pulmonar, uma vez que, na fase fetal, os pulmões ainda não estão funcionando para recebê-lo. É através desse canal que o sangue é direcionado para a aorta e, daí, para a artéria umbilical que, ligada à placenta, leva o sangue fetal para ser oxigenado e limpado dos resíduos do metabolismo fetal, votando depois novamente para o feto.
Ao nascer, logo que ocorre a laqueação do cordão umbilical, o sangue passa a ser oxigenado nos pulmões, e o canal arterial começa a ser fechado paulatinamente. A Persistência do Canal Arterial, depois dessa fase, caracteriza-se como uma cardiopatia congênita que permite um roubo de fluxo da aorta para as artérias pulmonares e, se não for corrigida, pode levar à hipertensão pulmonar, na fase adulta, que, em alguns casos, pode ser uma condição de gravidade.
Nos casos de certas cardiopatias congênitas, no entanto, a PCA é benigna. Esse é o caso do ventrículo único, da atresia da válvula tricúspede, e da transposição dos grandes vasos. Nesse caso, é benéfico que o canal permaneça aberto até que a cirurgia corretiva aconteça.
Sintomas
Os sintomas da PCA são mais presentes quando seu diâmetro é maior. Geralmente eles surgem associados aos quadros decorrentes da PCA, como a insuficiência do ventrículo esquerdo. Falta de apetite, irritabilidade, taquidispneia (respiração rápida e desconfortável) são alguns dos sintomas, assim como baixo ganho de peso, sudorese excessiva, infecções respiratórias superiores recidivantes e pneumonias.
Diagnóstico
Na ausculta cardíaca, percebe-se desde um sopro sistólico até um sopro contínuo típico dessa patologia. Normalmente um ecocardiograma define o diagnóstico mas, excepcionalmente, pode ser necessária uma angiotomografia ou angioressonância.
O que causa a doença
Embora a causa seja desconhecida em seus detalhes, cogita-se que o problema seja decorrente de mecanismos genéticos. A prematuridade do bebê, por interromper o desenvolvimento natural que levaria ao fechamento do canal arterial, é um fator de risco para a doença.
Cuidados
Nos prematuros normalmente o tratamento medicamentoso ou cirúrgico precoce são indicados. Caso não ocorra sucesso no fechamento, a criança precisará de medicações anticongestivas até que haja a correção percutânea (por meio de catetéres) ou cirúrgica da doença. Até que isso aconteça, a depender do quadro de comprometimento cardíaco, o paciente é afastado das atividades físicas.
Prevenção
É importante evitar condições que favoreçam o trabalho de parto prematuro, já que a prematuridade é um importante fator de risco. Caso haja histórico na família, é aconselhável realizar uma avaliação genética.
Tratamento
Em recém-nascidos, o tratamento é indicado quando existe excessivo fluxo de sangue para o coração e os pulmões, em decorrência dessa malformação. Caso o fechamento não ocorra espontaneamente, pode-se induzi-lo com medicação (indometacina, ibuprofeno), caso não haja contraindicação ao seu uso como insuficiência renal. No rol dos métodos poucos invasivos, está o cateterismo cardíaco, a depender da disponibilidade e anatomia da malformação. Em alguns casos, a cirurgia é indicada, principalmente na fase pré-escolar.
Prognóstico
O prognóstico é bom, uma vez que a malformação pode ser totalmente corrigida. Mas para que isso aconteça, é importante que o tratamento seja feito no tempo certo, antes que aconteça o comprometimento da musculatura do ventrículo esquerdo ou a ocorrência de hipertensão arterial pulmonar.
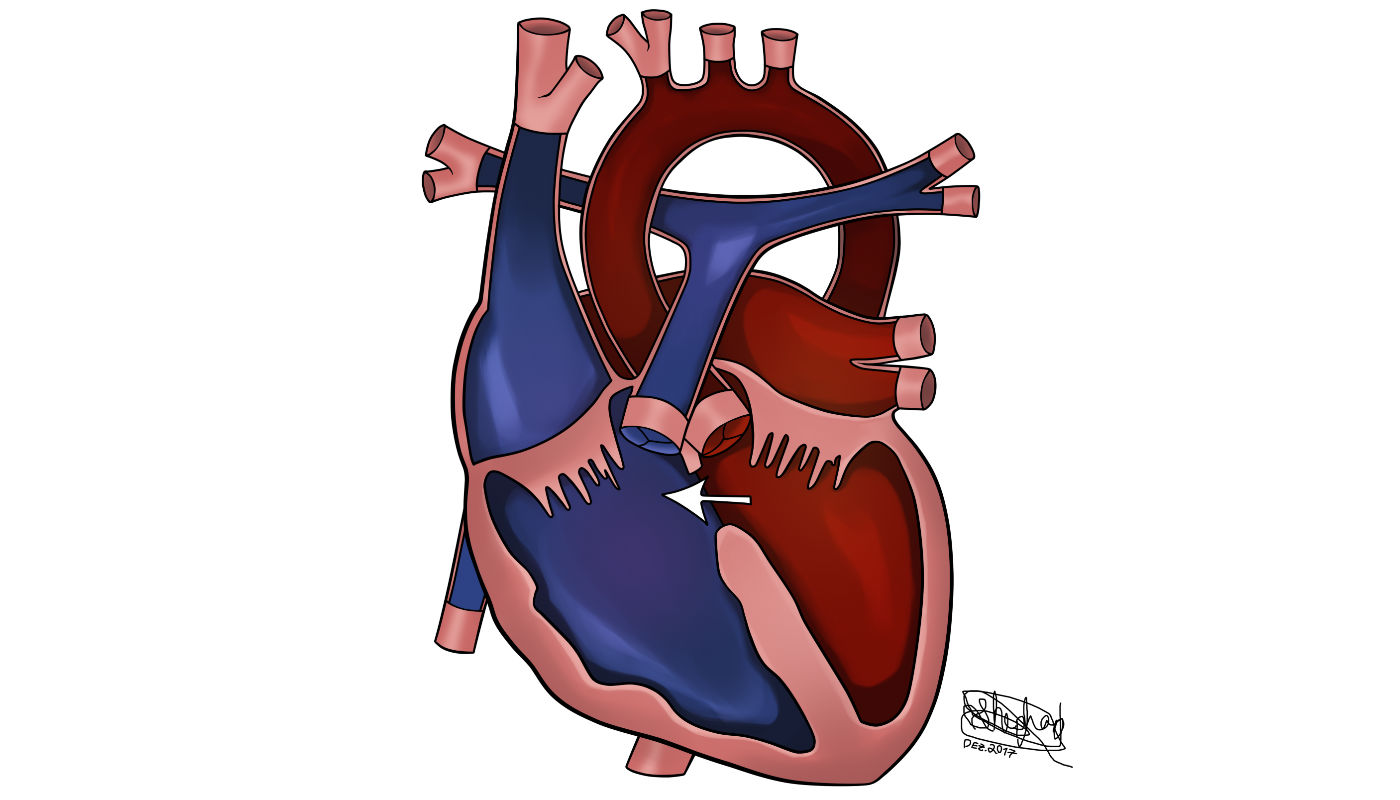
A transposição das grandes artérias é cerca de 3% de todas as cardiopatias congênitas, e cerca de 20% de todas as cardiopatias cianóticas.
Faixa etária:
Presente ao nascimento.
O que é a doença:
É uma discordância ventrículo arterial na qual a aorta emerge do o ventrículo direito e a artéria pulmonar do ventrículo esquerdo. O defeito anatômico da D-TGA leva a uma doença cianótica e como resultado há duas circulações paralelas. A primeira manda sangue desoxigenado para o átrio direito e de volta à circulação sistêmica via ventrículo direito e aorta. A segunda envia sangue oxigenado para o átrio esquerdo e de volta aos pulmões pelo ventrículo esquerdo e artéria pulmonar.
![]()
Ilustração por Izabella Hanada
Sintomas:
Cianose, taquipneia (respiração rápida) e pode ter pulsos reduzidos se houver coarctação ou interrupção da aorta.
Diagnóstico:
Devido às altas taxas de mortalidade neonatal e infantil, é importante fazer o diagnóstico de D- TGA o mais rápido possível, no pré-natal com o eco fetal, mas a maioria dos casos ainda é diagnosticada após o parto com eletrocardiograma, radiografia de tórax, ecocardiograma e cateterismo cardíaco.
O que causa a malformação:
Ao contrário de muitas outras formas de cardiopatia congênita, o D-TGA não está associado a nenhuma anormalidade genética comum.
Tratamento:
Antes da correção cirúrgica, alguns procedimentos médicos podem ser necessários como, por exemplo, a infusão de prostaglandin E1 (PGE1), tratamento com bicarbonato de sódio para corrigir a acidose metabólica, suporte de ventilação mecânica em casos de edema pulmonar e insuficiência respiratória e a septostomia atrial com balão que é um procedimento utilizado em pacientes com hipoxemia grave.
Os procedimentos cirúrgicos de Jatene e Rastelli são correções anatômicas cirúrgicas possíveis.
Por Izabella Hanada & Dra. Vanessa Guimarães – CREMESP 118.641
A Tetralogia de Fallot é a cardiopatia congênita cianótica mais comum e uma das primeiras corrigida por cirurgia.

O que é a doença
É uma alteração grave na formação do coração sustentada por 4 modificações: estenose pulmonar infundibular (um estreitamento da saída do fluxo sanguíneo do ventrículo direito – uma das “bombas” do coração, que manda sangue dele para o pulmão -, o que resulta em dificuldade dessa ejeção de sangue); hipertrofia do ventrículo direito (a parede muscular dessa cavidade engrossa, pelo aumento de pressão dentro dela); comunicação interventricular (uma fenda na parede que divide as “bombas” do coração e dextroposição da aorta, a qual fica desviada sobre o topo do septo interventricular, facilitando a ejeção de sangue pouco oxigenado, diretamente para a circulação do corpo.
Sintomas
O quadro mais frequente após o nascimento da criança é o de cianose (arroxeamento de lábios e leitos ungueais) leve a moderada. É daí que vem o termo de “bebê azul”!
Caso sejam graves os desalinhamentos, pode haver cianose grave associado a falta de ar intensa e pouco ganho de peso neonatal.
Tratamento
O principal objetivo do tratamento é regular os níveis de oxigênio no sangue das crianças. Nos quadros graves, será necessário iniciar Prostaglandina E 1 no berçário e então a cirurgia de Blalock-Taussig modificada ou a colocação de um stent no canal arterial após o nascimento. A cirurgia corretiva envolve: fechamento do septo interventricular com tela de Dacron para que o sangue flua corretamente; ressecção muscular para ampliação da via de saída do ventrículo direito, e valvoplastia pulmonar.
Prevenção
Como a etiologia da doença é fortemente genética, é importante o aconselhamento genético e o diagnóstico precoce pelo eco fetal.
Por Rafael Antunes & Dra. Vanessa Guimarães – CREMESP 118.641
A síndrome de Williams é uma condição genética que está presente no nascimento e pode afetar qualquer pessoa. É caracterizada por problemas médicos, incluindo doenças cardiovasculares, atrasos no desenvolvimento e dificuldades de aprendizagem. Tais questões médicas geralmente ocorrem lado a lado com habilidades verbais, personalidades altamente sociais e grande afinidade pela música.
A Síndrome de Williams ocorre igualmente em homens e mulheres e em todas as culturas em todo o mundo. É causada pela deleção espontânea, no óvulo ou no espermatozóide, de 26-28 genes no cromossomo 7 no momento da concepção. Na maioria das famílias, a criança com síndrome de Williams pode ser a única a ter essa condição clínica. A síndrome afeta 1 em 7.500 pessoas em todo o mundo. Crianças com síndrome de Williams tendem a ser sociais, amigáveis e cativantes, mas também encaram grandes desafios. Problemas cardiovasculares leves a potencialmente fatais, por exemplo, podem estar presentes no nascimento ou se tornarem evidentes enquanto bebês e crianças pequenas. As crianças normalmente precisam de cuidados médicos caros, contínuos e intervenções multiprofissionais precoces (como fonoaudiologia, fisioterapia e terapia ocupacional) para ajudar a superar atrasos no desenvolvimento.
À medida que crescem, as crianças têm dificuldades com questões específicas como relações espaciais, números, raciocínio abstrato e funções executivas, o que pode gerar desafios nas próprias tarefas diárias. Quando adultos, a maioria das pessoas com síndrome de Williams precisará de apoio domiciliar para vivenciar todo o seu potencial.
Uma boa notícia é que as oportunidades de emprego e moradia para indivíduos com síndrome de Williams estão aumentando e melhorando anualmente.
Devido à sua natureza amigável e necessidade de companhia, as oportunidades de interação social são importantes para aqueles com síndrome de Williams ao longo de suas vidas. No entanto, as pessoas com a síndrome geralmente não processam dicas sociais sutis, muitas vezes dificultando a formação de relacionamentos duradouros.
À medida que as pessoas com síndrome de Williams amadurecem, muitas vezes experimentam um isolamento intenso que pode levar ao aumento da ansiedade e depressão. Existem muitas características comuns às pessoas com síndrome de Williams. A deleção da elastina é responsável por muitas das características físicas. Outros problemas médicos e de desenvolvimento são provavelmente causados pelas deleções do material genético adicional próximo ao gene da elastina no cromossomo 7. No entanto, o número de características presentes e quais características estão presentes varia de criança para criança, embora mais de 95% dos indivíduos tenham uma deleção genética idêntica de 26-28.
O diagnóstico da síndrome de Williams geralmente é realizado em duas partes: Diagnóstico clínico baseado em uma variedade de características. Confirmação de teste médico/genético através de um teste de DNA realizado em uma pequena quantidade de sangue do indivíduo. Praticamente todas as pessoas (98-99%) com características típicas da síndrome de Williams terão deleção do gene da elastina. Em termos mais técnicos: a síndrome de Williams é o resultado de uma deleção da região 7q11.23 do cromossomo 7, contendo 26-28 genes, incluindo o gene da elastina. A elastina é o “gene marcador” da síndrome de Williams. A síndrome de Williams é uma síndrome de genes contíguos, o que significa que todos os genes deletados “se alinham” dentro da “região crítica” da síndrome de Williams de 26 a 28 genes. Existem dois testes de DNA que podem determinar se uma pessoa tem síndrome de William: o Nb teste FISH e o Microarray. É importante enfatizar que a síndrome é um diagnóstico genético e um indivíduo sem a deleção do gene não tem síndrome de Williams (ou seja, uma pessoa que foi diagnosticada clinicamente com a síndrome com base nas características fenotípicas, mas posteriormente fez o teste genético e não tem a deleção na verdade NÃO tem síndrome de Williams.
É extremamente improvável que qualquer outro membro da família também tenha síndrome de Williams. Isso ocorre porque a exclusão ocorre espontaneamente espontaneamente a uma taxa de apenas 1 em 7.500. No entanto, se o indivíduo com síndrome de Williams planeja se tornar pai, há uma chance de 50/50 de que seu filho tenha síndrome de Williams. Se tal situação ocorrer, deve-se consultar um obstetra sobre o uso do teste FISH para testes pré-natais em células embrionárias.
FISH é um tipo de análise cromossômica especializada que utiliza sondas de elastina especialmente preparadas. Se um paciente tem 2 cópias do gene da elastina (uma em cada um de seus cromossomos 7), provavelmente não tem a síndrome. Se o indivíduo tiver apenas uma cópia, o diagnóstico da síndrome está confirmado.
Intervenções Terapêuticas
Quase todos os indivíduos com síndrome de Williams se beneficiarão de intervenções terapêuticas para ajudar a superar atrasos no desenvolvimento, problemas articulares, problemas motores finos e outras características comuns à síndrome de Williams. O médico pode ajudar a determinar a necessidade de terapia ou fornecer um encaminhamento para que o paciente seja avaliado por um profissional de estratégias terapêuticas. É importante iniciar as terapias o mais rápido possível para obter o máximo benefício. A maioria das crianças em idade escolar pode receber intervenções terapêuticas como parte de suas atividades. As escolas participam fornecendo terapia para superar as dificuldades que forem um “bloqueio” para a educação bem-sucedida de uma criança. Para alguns atrasos, os serviços particulares são a única saída. Existem materiais e fichas informativas terapêuticas que foram criados por terapeutas com profundo conhecimento sobre a síndrome de Williams.
FISIOTERAPIA
Crianças com síndrome de Williams podem ter problemas de equilíbrio e tônus muscular. A fisioterapia abordará o desenvolvimento de habilidades motoras grossas em crianças com síndrome de Williams.
TERAPIA OCUPACIONAL
As crianças com síndrome de Williams geralmente apresentam déficits visuais-espaciais e dificuldade no controle muscular fino, como pegar pequenos objetos. A terapia ocupacional aborda principalmente o desenvolvimento de habilidades motoras finas em crianças com síndrome de Williams, mas também importante fonte de ajuda para problemas de alimentação e sensibilidade a texturas.
FONOAUDIOLOGIA
O início da fala geralmente é atrasado em crianças com síndrome de Williams e a articulação das palavras pode ser afetada por problemas de tônus muscular. Uma vez que a fala tenha sido adquirida, as crianças muitas vezes experimentam dificuldades com o processamento de informações. Um fonoaudiólogo pode resolver todos os problemas relacionados à fala/linguagem.
MUSICOTERAPIA
A musicoterapia está ganhando espaço em muitas escolas nacionalmente, e a ligação direta entre a síndrome de Williams e a musicalidade pode vir a ajudar. A musicoterapia envolve ensinar e reforçar outras habilidades cognitivas e físicas através do uso da música. A musicoterapia NÃO depende da habilidade musical e NÃO ensina as crianças a tocar um instrumento. Ela utiliza o amor natural de uma criança pela música para ajudá-la a melhorar outras tarefas.
OUTROS TIPOS DE TERAPIAS
Pacientes com síndrome de Williams se beneficiam de muitos tipos diferentes de suporte terapêutico. Além da terapia ocupacional, física e fonoaudiológica, as crianças com síndrome de Williams geralmente participam e obtêm excelentes benefícios de tipos menos tradicionais de terapia – especialmente musicoterapia, equoterapia e equitação terapêutica e terapias baseadas em som.
EQUOTERAPIA/HIPOTERAPIA
A equoterapia pode trazer benefícios para crianças com síndrome de Williams, como acontece com qualquer criança com deficiência. “Hipoterapia” é um termo que se refere ao uso do movimento do cavalo como uma ferramenta de tratamento por fisioterapeutas, terapeutas ocupacionais e fonoaudiólogos para tratar deficiências, limitações funcionais e deficiências em pacientes com disfunção neuromusculoesquelética. A equoterapia é usada como parte de um programa de tratamento integrado para alcançar resultados funcionais.” A equitação terapêutica aborda também a autoestima e o bem-estar emocional.
TERAPIAS BASEADAS EM SOM
Terapias baseadas em som provaram ser úteis para algumas crianças com síndrome de Williams. Existem várias formas de terapias baseadas em som disponíveis. Uma dela é a Terapia de Integração Auditiva – originalmente projetada para indivíduos com autismo; mas já usada para indivíduos com dislexia e transtornos de déficit de atenção e hiperatividade. O objetivo é reduzir a hipersensibilidade aos sons e remediar as dificuldades auditivas.